Syndrome de Gilbert : symptômes et traitement

Selon les estimations effectuées, le syndrome de Gilbert est une condition présente chez 1 % de la population mondiale. Néanmoins, il se présente à une moyenne d’environ 5 à 10 % chez certaines populations. Il s’agit d’un problème héréditaire qui se détecte normalement chez les adultes d’entre 20 et 30 ans.
Le plus fréquent est que le syndrome de Gilbert se détecte lorsque l’on fait une prise de sang de routine. Cette maladie a été décrite pour la première fois en 1902 par Augustin Nicolas Gilbert et Pierre Lereboullet. D’où le nom du syndrome.
Le syndrome de Gilbert est une condition bénigne mais chronique. Il est dû à une mutation génétique héréditaire. Beaucoup des personnes qui souffrent de cette maladie l’ignorent car elle ne provoque pas un cadre de symptômes facilement détectable.
Le syndrome de Gilbert
Le syndrome de Gilbert se caractérise par des niveaux élevés de bilirubine. Même si elle peut générer une préoccupation, cette condition est considérée comme bénigne.
Le syndrome de Gilbert est une condition héréditaire qui se caractérise par des niveaux élevés de bilirubine dans le sang. On le connaît aussi sous le nom d’insuffisance hépatique constitutionnelle et d’ictère familial non hémolytique.
Il s’agit d’un dysfonctionnement au cours duquel le foie ne traite pas la bilirubine de la façon dont il devrait le faire. Néanmoins, le syndrome de Gilbert ne produit pas de dommages. Il faut préciser que la bilirubine est une substance qui vient des globules rouges qui vieillissent et sont traités par le foie.
Normalement, la bilirubine est évacuée de l’organisme à travers la matière fécale et l’urine. Lorsque la bilirubine grimpe, le foie ne parvient pas à la traiter de façon adéquate. À cause de cela, cette substance commence à être absorbée par la peau et les tissus.
Découvrez aussi : Le cancer est-il héréditaire ? Données à connaître
Causes
Le syndrome de Gilbert est la conséquence de la réduction de la capacité des hépatocytes (cellules du foie) à excréter la bilirubine. Lorsque les globules rouges vieillissent et se détériorent, ils libèrent de l’hémoglobine. Celle-ci est métabolisée et forme un groupe de substances appelées « Hémo ».
L’hémo se transforme d’abord en biliverdine et, ensuite, en sorte de première version de bilirubine, dite « non conjuguée » ou indirecte. Quand celle-ci passe par le foie et se combine à l’acide glucuronique, une réaction a lieu et aboutit à la bilirubine directe ou conjuguée.
Si le processus se fait de façon adéquate, la bilirubine devient soluble dans l’eau et est excrétée par la bile. Néanmoins, chez certaines personnes, on retrouve un gène muté qui empêche la production normale de l’enzyme régulant la production de bilirubine. Ceci conduit au syndrome de Gilbert.
Symptômes et diagnostic du syndrome de Gilbert
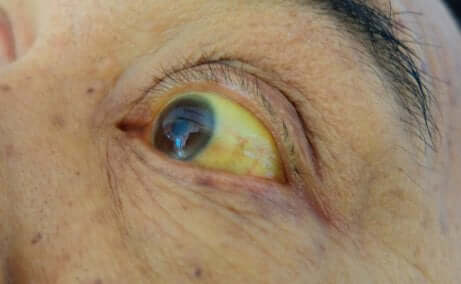
L’ictère est la coloration jaunâtre de la peau et des yeux. C’est le symptôme le plus caractéristique du syndrome de Gilbert.
Le principal symptôme du syndrome de Gilbert est l’ictère, c’est-à-dire la couleur jaunâtre de la peau et des yeux. Il devient plus visible dans des moments de grand stress, quand il y a un jeûne prolongé, en cas de maladie infectieuse ou quand on fait de grands efforts.
Un autre des symptômes présents est l’incapacité à assimiler correctement certaines substances, et surtout certains médicaments. Plus concrètement, certaines personnes touchées par ce syndrome présentent une augmentation du risque de toxicité au paracétamol.
Il n’y a pas d’autres symptômes reconnaissables. Lors de l’analyse de sang des personnes atteintes de cette maladie, la bilirubine indirecte présente des taux élevés, tandis que la bilirubine directe reste à des taux normaux. Les analyses hépatiques sont également normales et on ne détecte aucune anomalie lors des examens d’imagerie.
Découvrez aussi : Les maladies du foie
Traitement
Le syndrome de Gilbert n’exige aucun type de traitement car il s’agit d’une anomalie qui ne produit pas de dommages. On le considère donc comme bénin. Ce dysfonctionnement ne provoque aucun type d’incidence sur la qualité de vie d’une personne, à tel point qu’il passe très souvent inaperçu.
Aucun type de complications n’a été recensé suite à ce syndrome, et il n’exige aucun régime alimentaire spécial. Il n’affecte pas non plus la capacité à faire de l’exercice de façon normale. Il faut juste être attentif à l’ingestion de certains médicaments comme le paracétamol.
Comme nous l’avons souligné, il existe, avec le syndrome de Gilbert, un risque augmenté d’effets hépatiques secondaires suite à l’ingestion de certains médicaments. Pour le reste, une personne avec cette condition peut mener une vie parfaitement normale, sans avoir besoin de soins spéciaux.
L’ictère peut apparaître et disparaître plusieurs fois au cours de la vie, sans impliquer le moindre risque pour la santé. Une fois le diagnostic établi, aucun type de suivi médical n’est nécessaire pour cette maladie.
Toutes les sources citées ont été examinées en profondeur par notre équipe pour garantir leur qualité, leur fiabilité, leur actualité et leur validité. La bibliographie de cet article a été considérée comme fiable et précise sur le plan académique ou scientifique
- Delgado Peña, Y., Fleta Zaragozano, J., Lázaro Almarza, A., Gracía Casanova, M., Jiménez Vidal, A., & Olivares López, J. (2007). Síndrome de Gilbert: estudio de 12 observaciones y revisión de la bibliografía médica. Acta Pediátrica Española, 65(8), 404-408.
- Thoguluva Chandrasekar V, John S. Gilbert Syndrome. [Updated 2019 Apr 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470200/
- Qian JD, Hou FQ, Wang TL, Shao C, Wang GQ. Gilbert syndrome combined with prolonged jaundice caused by contrast agent: Case report. World J Gastroenterol. 2018;24(13):1486–1490. doi:10.3748/wjg.v24.i13.1486
- Wagner, K. H., Shiels, R. G., Lang, C. A., Seyed Khoei, N., & Bulmer, A. C. (2018, February 17). Diagnostic criteria and contributors to Gilbert’s syndrome. Critical Reviews in Clinical Laboratory Sciences. Taylor and Francis Ltd. https://doi.org/10.1080/10408363.2018.1428526
Ce texte est fourni à des fins d'information uniquement et ne remplace pas la consultation d'un professionnel. En cas de doute, consultez votre spécialiste.







